Enhanced spin Hall conductivity and charge to spin conversion efficiency in strained orthorhombic SnSe through orbital selective hybridization
E. Ketkar, Gaurav K. Shukla, Seung-Cheol Lee, Satadeep Bhattacharjee, Sanjay Singh
Applied Physics Letters
Abstract
Enhanced spin Hall conductivity and charge to spin conversion efficiency in strained orthorhombic SnSe through orbital selective hybridization
The realization of the spin Hall effect has opened new frontiers for the design of efficient memory storage devices facilitated by the conversion of charge currents to spin currents. Here, using the Kubo formula, we calculate the intrinsic spin Hall conductivity (SHC) of orthorhombic tin selenide (o-SnSe) under the influence of isotropic compressive strain in the ab-plane. As the strain is gradually increased, we obtain a substantial hybridization between the pz orbitals of Sn and Se atoms of an electron pocket from the lowest conduction band and the topmost valence band, respectively. This hybridization process greatly enhances the SHC at the Fermi level and charge-to-spin conversion efficiency, the latter of which is superior to that of popular transition metals such as Ta and Pt. This makes strained o-SnSe an attractive candidate for use in spintronic devices.
84
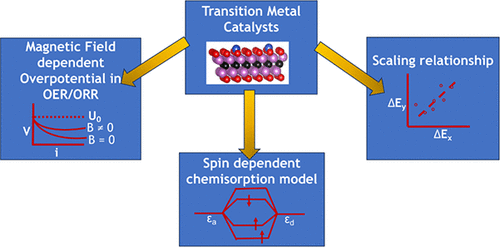
Insights into Heterogeneous Catalysis on Surfaces with 3d Transition Metals: Spin-Dependent Chemisorption Models and Magnetic Field Effects
Satadeep Bhattacharjee, Swetarekha Ram, and Seung-Cheol Lee
The Journal of Physical Chemistry Letters
Abstract
Insights into Heterogeneous Catalysis on Surfaces with 3d Transition Metals: Spin-Dependent Chemisorption Models and Magnetic Field Effects
This Perspective provides an overview of recent developments in the field of 3d transition metal (TM) catalysts for different reactions, including oxygen-based reactions such as the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER). The spin moments of 3d TMs can be exploited to influence chemical reactions, and recent advances in this area, including the theory of chemisorption based on spin-dependent d-band centers and magnetic field effects, are discussed. The Perspective also explores the use of scaling relationships and surface magnetic moments in catalyst design as well as the effect of magnetism on chemisorption and vice versa. In addition, recent studies on the influence of a magnetic field on the ORR and the OER are presented, demonstrating the potential of ferromagnetic catalysts to enhance these reactions through spin polarization.
83
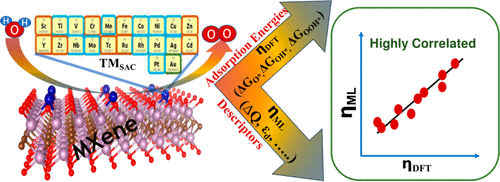
Combining First-Principles Modeling and Symbolic Regression for Designing Efficient Single-Atom Catalysts in the Oxygen Evolution Reaction on Mo2CO2 MXenes
Swetarekha Ram , Gwan Hyun Choi, Albert S. Lee, Seung-Cheol Lee, and Satadeep Bhattacharjee
ACS Applied Materials & Interfaces
Abstract
Combining First-Principles Modeling and Symbolic Regression for Designing Efficient Single-Atom Catalysts in the Oxygen Evolution Reaction on Mo2CO2 MXenes
In this study, we address the significant challenge of overcoming limitations in the catalytic efficiency for the oxygen evolution reaction (OER). The current linear scaling relationships hinder the optimization of the electrocatalytic performance. To tackle this issue, we investigate the potential of designing single-atom catalysts (SACs) on Mo2CO2 MXenes for electrochemical OER using first-principles modeling simulations. By employing the Electrochemical Step Symmetry Index (ESSI) method, we assess OER intermediates to fine-tune the activity and identify the optimal SAC for Mo2CO2 MXenes. Our findings reveal that both Ag and Cu exhibit effectiveness as single atoms for enhancing OER activity on Mo2CO2 MXenes. However, among the 21 chosen transition metals (TMs) in this study, Cu stands out as the best catalyst for tweaking the overpotential (ηOER). This is due to Cu’s lowest overpotential compared to other TMs, which makes it more favorable for the OER performance. On the other hand, Ag is closely aligned with ESSI = ηOER, making the tuning of its overpotential more challenging. Furthermore, we employ symbolic regression analysis to identify the significant factors that exhibit a correlation with the OER overpotential. By utilizing this approach, we derive mathematical formulas for the overpotential and identify key descriptors that affect the catalytic efficiency in the electrochemical OER on Mo2CO2 MXenes. This comprehensive investigation not only sheds light on the potential of MXenes in advanced electrocatalytic processes but also highlights the prospect of improved activity and selectivity in OER applications.
82
High-frequency complex impedance analysis of the two-dimensional semiconducting MXene Ti_2CO_2
Anup Kumar Mandia, Rohit Kumar, Namitha Anna Koshi, Seung-Cheol Lee, Satadeep Bhattacharjee and Bhaskaran Muralidharan
Physica Scripta
Abstract
High-frequency complex impedance analysis of the two-dimensional semiconducting MXene Ti_2CO_2
The two-dimensional compound group of MXenes, which exhibit unique optical, electrical, chemical, and mechanical properties, are an exceptional class of transition metal carbides and nitrides. In addition to traditional applications in Li-S, Li-ion batteries, conductive electrodes, hydrogen storage, and fuel cells, the low lattice thermal conductivity coupled with high electron mobility in the semiconducting oxygen-functionalized MXene Ti2CO2 has led to the recent interests in high-performance thermoelectric and nanoelectronic devices. Apart from the above dc- transport applications, it is crucial to also understand ac- transport across them, given the growing interest in applications surrounding wireless communications and transparent conductors. In this work, we investigate using our recently developed abinitio transport model, the real and imaginary components of electron mobility and conductivity to conclusively depict carrier transport beyond the room temperature for frequency ranges upto the terahertz range. We also contrast the carrier mobility and conductivity with respect to the Drude's model to depict its inaccuracies for a meaningful comparison with experiments. Our calculations show the effect of acoustic deformation potential scattering, piezoelectric scattering, and polar optical phonon scattering mechanisms. Without relying on experimental data, our model requires inputs calculated from first principles using density functional theory. Our results set the stage for providing abinitio based ac- transport calculations given the current research on MXenes for high-frequency applications.
81
Spin torques and anomalous velocity in spin textures induced by fast electron injection from topological ferromagnets: The role of gauge fields
Satadeep Bhattacharjee, Seung-Cheol Lee
Journal of Physics: Condensed Matter
Abstract
Spin torques and anomalous velocity in spin textures induced by fast electron injection from topological ferromagnets: The role of gauge fields
A new method for analyzing magnetization dynamics in spin textures under the influence of fast electron injection from topological ferromagnetic sources such as Dirac half metals has been proposed. These electrons, traveling at a velocity v with a non-negligible value of $v/c$ (where c is the speed of light), generate a non-equilibrium magnetization density in the spin-texture region, which is related to an electric dipole moment via relativistic interactions. When this resulting dipole moment interacts with gauge fields in the spin-texture region, an effective field is created that produces spin torques. These torques, like spin-orbit torques that occur when electrons are injected from a heavy metal into a ferromagnet, can display both damping-like and anti-damping-like properties. Finally, we demonstrate that such an interaction between the dipole moment and the gauge field introduces an anomalous velocity that can contribute to transverse electrical conductivity in the spin texture in a way comparable to the topological Hall effect.
Despite recent advances in colloidal quantum dot (CQD) photovoltaics, several challenges persist and hinder further improvements. In particular, the Fermi level mismatch between the iodide-treated photoactive and thiol-treated hole-transporting CQD layers creates an unfavorable energy band for hole collection. Furthermore, the numerous surface cracks in the thiol-treated CQD layer facilitate direct contact between the photoactive CQD layer and the metal electrode, consequently leading to reduced device performance. To address these issues, a polycatechol functionalized MXene (PCA-MXene) that can serve both as a dopant and an interlayer for CQD photovoltaics is developed. By achieving a uniformly dispersed mixture in a butylamine solvent, PCA-MXene enables the effective combination of MXene and CQDs. This results in the modification of the work function of CQDs and the modulation of the energy band alignment, ultimately promoting enhanced hole extraction. Moreover, the PCA-MXene employed as an interlayer effectively covers the surface cracks present in the thiol-treated CQD layer. This coverage inhibits both metal electrode penetration and moisture intrusion into the device. Owing to these advantages, the CQD photovoltaics incorporating PCA-MXene achieve a power conversion efficiency (PCE) of 13.6%, accompanied by enhanced thermal stability, in comparison to the reference device with a PCE of 12.8%.
79
CrysGNN: Distilling Pre-trained Knowledge to Enhance Property Prediction for Crystalline Materials
Proceedings of the AAAI Conference on Artificial Intelligence
Abstract
CrysGNN: Distilling Pre-trained Knowledge to Enhance Property Prediction for Crystalline Materials
In recent years, graph neural network (GNN) based approaches have emerged as a powerful technique to encode complex topological structure of crystal materials in an enriched repre- sentation space. These models are often supervised in nature and using the property-specific training data, learn relation- ship between crystal structure and different properties like formation energy, bandgap, bulk modulus, etc. Most of these methods require a huge amount of property-tagged data to train the system which may not be available for different prop- erties. However, there is an availability of a huge amount of crystal data with its chemical composition and structural bonds. To leverage these untapped data, this paper presents CrysGNN, a new pre-trained GNN framework for crystalline materials, which captures both node and graph level structural information of crystal graphs using a huge amount of unla- belled material data. Further, we extract distilled knowledge from CrysGNN and inject into different state of the art prop- erty predictors to enhance their property prediction accuracy. We conduct extensive experiments to show that with distilled knowledge from the pre-trained model, all the SOTA algo- rithms are able to outperform their own vanilla version with good margins. We also observe that the distillation process provides significant improvement over the conventional ap- proach of finetuning the pre-trained model. We will release the pre-trained model along with the large dataset of 800K crys- tal graph which we carefully curated; so that the pre-trained model can be plugged into any existing and upcoming models to enhance their prediction accuracy.
78
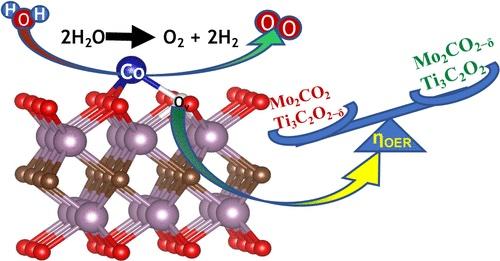
Unraveling the Influence of Oxygen Vacancies on the OER Performance of Co Single-Atom Catalysts Adsorbed on MXenes
Swetarekha Ram , Gwan Hyun Choi, Albert S. Lee, Seung-Cheol Lee, and Satadeep Bhattacharjee
The Journal of Physical Chemistry C
Abstract
Unraveling the Influence of Oxygen Vacancies on the OER Performance of Co Single-Atom Catalysts Adsorbed on MXenes
To enable efficient energy conversion and storage, the development of effective electrocatalysts for the oxygen evolution reaction (OER) is crucial. Single-atom catalysts (SACs) with 100% active sites for the OER are highly promising in this regard. In this study, we investigated the OER activities of Co single atoms (CoSA) adsorbed on metallic MXenes, including Ti3C2O2 and Mo2CO2, both in their stoichiometric form and with oxygen vacancies (Ov), using spin-polarized first-principles-based calculations. The rate-determining step in each case was found to be the conversion of *O from *OH. Our calculations showed that the presence of oxygen vacancies decreased the OER activity in CoSA@Ti3C2O2-δ, resulting in a higher overpotential, while it increased the OER activity in CoSA@Mo2CO2. We explain such results using insights from the density of states, charge density variation, and bonding analysis via crystal orbital Hamilton population. We also show that the hybridization between the d-states of the CoSA and the transition metal sites of the catalyst-bed (Ti/Mo) plays a decisive role.
77
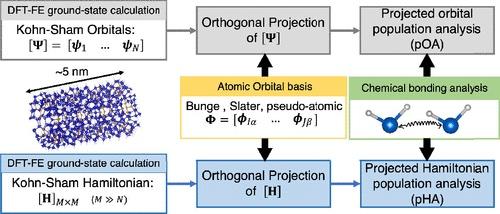
Chemical Bonding in Large Systems Using Projected Population Analysis from Real-Space Density Functional Theory Calculations
Kartick Ramakrishnan, Sai Krishna Kishore Nori, Seung-Cheol Lee, Gour P. Das, Satadeep Bhattacharjee, and Phani Motamarri
Journal of Chemical Theory and Computation
Abstract
Chemical Bonding in Large Systems Using Projected Population Analysis from Real-Space Density Functional Theory Calculations
We present an efficient and scalable computational approach for conducting projected population analysis from real-space finite-element (FE)-based Kohn–Sham density functional theory calculations (DFT-FE). This work provides an important direction toward extracting chemical bonding information from large-scale DFT calculations on materials systems involving thousands of atoms while accommodating periodic, semiperiodic, or fully nonperiodic boundary conditions. Toward this, we derive the relevant mathematical expressions and develop efficient numerical implementation procedures that are scalable on multinode CPU architectures to compute the projected overlap and Hamilton populations. The population analysis is accomplished by projecting either the self-consistently converged FE discretized Kohn–Sham orbitals or the FE discretized Hamiltonian onto a subspace spanned by a localized atom-centered basis set. The proposed methods are implemented in a unified framework within the DFT-FE code where the ground-state DFT calculations and the population analysis are performed on the same FE grid. We further benchmark the accuracy and performance of this approach on representative material systems involving periodic and nonperiodic DFT calculations with LOBSTER, a widely used projected population analysis code. Finally, we discuss a case study demonstrating the advantages of our scalable approach to extract the quantitative chemical bonding information of hydrogen chemisorbed in large silicon nanoparticles alloyed with carbon, a candidate material for hydrogen storage.
76
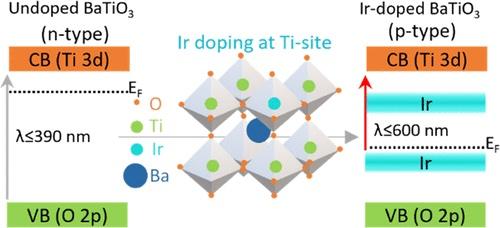
Iridium-Doping as a Strategy to Realize Visible-Light Absorption and p-Type Behavior in BaTiO3
Sujana Chandrappa, Simon Joyson Galbao, P. S. Sankara Rama Krishnan, Namitha Anna Koshi, Srewashi Das, Stephen Nagaraju Myakala, Seung-Cheol Lee, Arnab Dutta, Alexey Cherevan, Satadeep Bhattacharjee, and Dharmapura H. K. Murthy
The Journal of Physical Chemistry C
Abstract
Iridium-Doping as a Strategy to Realize Visible-Light Absorption and p-Type Behavior in BaTiO3
BaTiO3 (BTO) typically demonstrates a strong n-type character with absorption only in the ultraviolet (λ ≤ 390 nm) region. Extending the applications of BTO to a range of fields necessitates a thorough insight into how to tune its carrier concentration and extend the optical response. Despite significant progress, simultaneously inducing visible-light absorption with a controlled carrier concentration via doping remains challenging. In this work, a p-type BTO with visible-light (λ ≤ 600 nm) absorption is realized via iridium (Ir) doping. Detailed analysis using advanced spectroscopy/microscopy tools revealed mechanistic insights into the n- to p-type transition. The computational electronic structure analysis further corroborated this observation. This complementary data helped establish a correlation between the occupancy and the position of the dopant in the band gap with the carrier concentration. A decrease in the Ti3+ donor-level concentration and the mutually correlated oxygen vacancies upon Ir doping is attributed to the p-type behavior. Due to the formation of Ir3+/Ir4+ in-gap energy levels within the forbidden region, the optical transition can be elicited from or to such levels, resulting in visible-light absorption. This newly developed Ir-doped BTO is a promising semiconductor with imminent applications in solar fuel generation and optoelectronics.